
Obtenir le marquage CE : Étapes, obligations et conformité
Guide complet marquage CE : définition, directives applicables, étapes d'obtention, dossier technique. Commercialisez vos produits en Europe.
Votre prototype fonctionne, les premiers tests sont prometteurs. Mais à quelques mois du lancement, vous découvrez que les matériaux ne sont pas certifiés, que la traçabilité n'a pas été prévue, et que la documentation ne répond pas aux exigences. Résultat : refonte complète, validation à reprendre, plusieurs mois de retard.
Cette situation est le cas d’école de l’erreur qui consiste à traiter le contrôle qualité et la conformité réglementaire comme des formalités de dernière minute.
Or, la norme ISO 13485 est un système qui, lorsqu'il est intégré dès la conception, guide le développement et sécurise la mise sur le marché de votre produit. Cet article vous explique concrètement comment transformer cette norme en atout pour développer des dispositifs médicaux conformes, industrialisables et prêts à conquérir les marchés réglementés.
Pour bien comprendre les enjeux, commençons par définir précisément ce qu'est l'ISO 13485. Il s'agit d'une norme internationale qui spécifie les exigences pour un système de management de la qualité (SMQ) spécifique aux dispositifs médicaux. Son périmètre couvre l'ensemble du cycle de vie produit : conception, développement, production, stockage, distribution, installation et prestations associées.
Contrairement à des normes qualités plus généralistes comme l'ISO 9001, l'ISO 13485 est spécialement adaptée au domaine médical, avec un accent renforcé sur la traçabilité de chaque composant, la gestion formalisée des risques et des exigences documentaires strictes.
Cette spécificité sert à garantir que les dispositifs médicaux répondent de manière constante aux exigences des clients et aux exigences réglementaires. De plus, la norme est alignée avec les exigences du règlement (UE) 2017/745 relatif aux dispositifs médicaux (MDR), applicable en France et dans l’ensemble de l’Union européenne pour l’obtention du marquage CE. Elle est également partiellement reconnue par la Food and Drug Administration (FDA) aux États-Unis, dans la mesure où le système qualité américain est désormais largement harmonisé avec l’ISO 13485, sous réserve d’exigences réglementaires spécifiques supplémentaires
En premier lieu, les fabricants industriels de dispositifs médicaux de toutes classes (I, IIa, IIb, III). Mais l'obligation ne s'arrête pas là. Vos fournisseurs sont également concernés : fabricant de composants électroniques, prestataire de stérilisation, atelier d'usinage.
Les organismes prestataires intervenant dans la chaîne de valeur sont aussi visés : bureaux d'études, laboratoires d'essais, entreprises de maintenance.
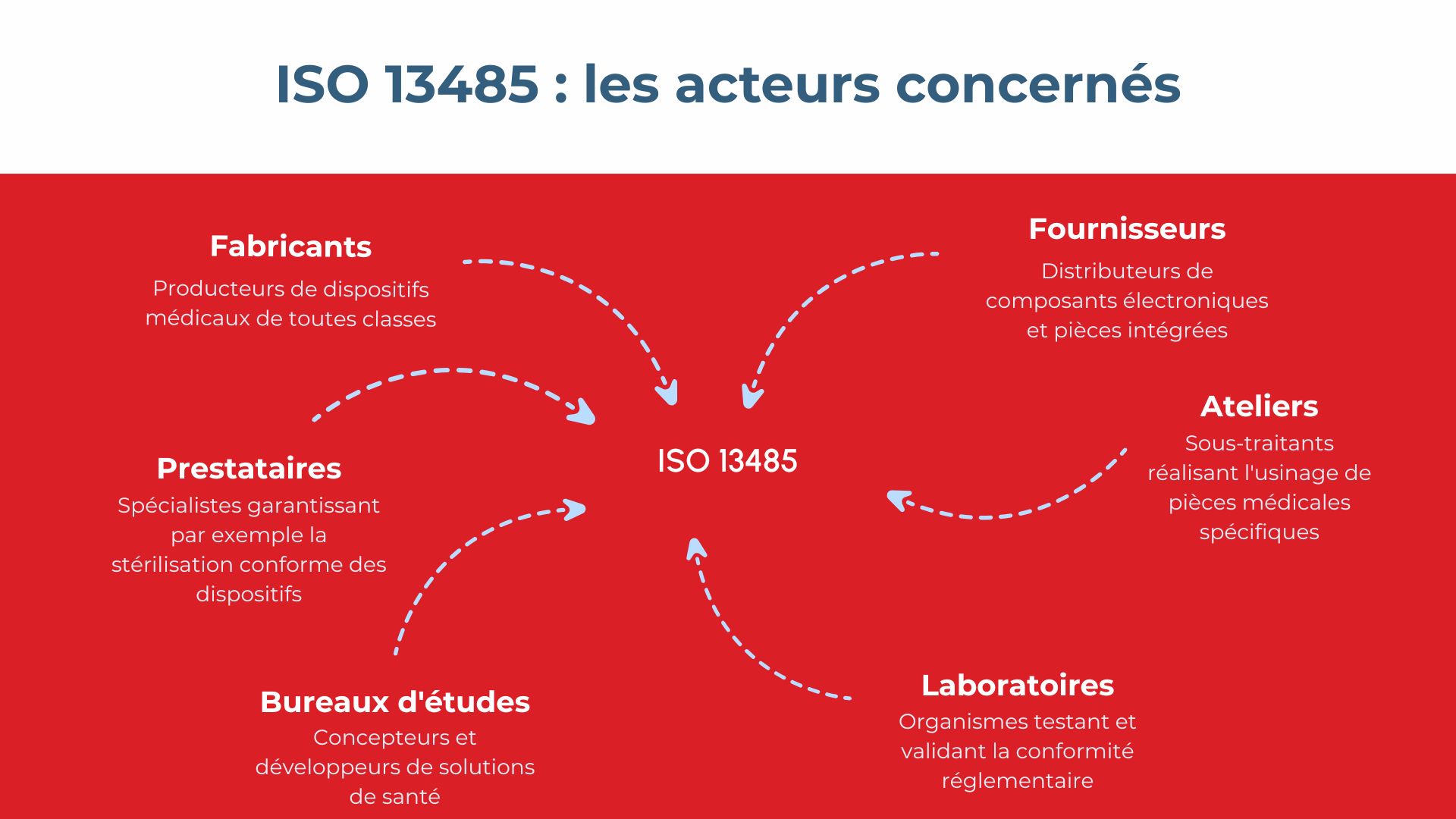
En bref, si vous participez à la chaîne de valeur d'un dispositif médical, l'ISO 13485 vous concerne.
Cette exigence peut sembler contraignante au premier abord, mais elle constitue en réalité votre meilleure assurance en cas de problème.
Concrètement, cela se traduit par deux corpus documentaires complémentaires. D'un côté, la documentation de conception et développement documente toutes les phases de développement : spécifications, choix techniques, tests, décisions.
De l'autre, le dossier technique du dispositif rassemble les éléments de conformité (spécifications finales, dessins, analyses de risques, rapports de validation, certificats matériaux).
Mais la traçabilité va encore plus loin. Chaque composant, chaque lot de production, chaque modification doit pouvoir être retracé. Pourquoi cette rigueur ? En cas de défaut détecté, la traçabilité peut permettre d'identifier rapidement les dispositifs concernés — parfois en quelques jours — plutôt que de rappeler l'ensemble de la production.
La gestion des modifications est elle aussi formalisée. Tout changement, même mineur, doit être documenté, évalué en termes de risques, testé et validé avant mise en œuvre. Cette rigueur évite les dérives progressives qui fragilisent le produit sans qu'on s'en aperçoive.
Au-delà de la documentation, le deuxième pilier fondamental de l'ISO 13485 concerne la gestion des risques formalisée, généralement mise en œuvre selon la norme ISO 14971. Contrairement à une idée reçue, cette approche n'est pas une étape finale du projet, mais un fil conducteur qui traverse tout le développement.
En pratique, l'analyse des risques se déroule tout au long du cycle de vie du produit. Elle démarre dès la phase de conception et se poursuit jusqu'à la fin de vie du dispositif, s'adaptant à chaque nouvelle information.
Cette méthodologie consiste d'abord à identifier les dangers potentiels :
Une fois ces dangers recensés, ils sont évalués et hiérarchisés, souvent à l'aide d'une approche croisant probabilité et gravité. Enfin, pour chaque risque significatif, des mesures de réduction sont définies : modification de conception, ajout de protections techniques, information dans la notice.
L'essentiel est de comprendre que l'analyse de risques doit influencer directement les choix de conception, pas simplement les documenter après coup. Prenons l'exemple d'un dispositif portable avec batterie. L'analyse identifie un risque de surcharge. Les mesures qui en découlent incluent alors : intégration d'un circuit de protection, sélection d'une batterie certifiée, mise en place de tests en conditions extrêmes.
La conformité se prouve par des tests rigoureux et documentés.
D'un côté, la vérification de conception répond à la question : "Avons-nous construit le produit correctement ?" Elle confirme que le produit répond aux spécifications d'entrée définies. Cette vérification s'appuie sur des tests, des calculs, des inspections qui démontrent objectivement la conformité aux spécifications.
De l'autre, la validation de conception répond à une question plus large : "Avons-nous construit le bon produit ?" Elle démontre que le produit répond aux besoins des utilisateurs dans les conditions réelles d'usage. À cette fin, la validation peut nécessiter des études cliniques, des tests sur patients ou utilisateurs finaux.
Enfin, pour compléter ce dispositif de preuve, la validation de processus intervient au niveau de la production. Elle prouve que les processus de fabrication produisent de manière répétable des résultats conformes. Cette étape est particulièrement critique pour les procédés spéciaux dont le résultat ne peut pas être entièrement vérifié par inspection : soudure, stérilisation, traitement thermique.
Toutes ces étapes exigent une documentation obligatoire : protocoles de tests détaillés, rapports complets incluant les résultats bruts, analyses statistiques démontrant la reproductibilité.

Les décisions prises en phase amont conditionnent directement la facilité de certification en aval. Autrement dit, intégrer la norme ISO 13485 dès le départ permet de limiter fortement les refontes coûteuses en phase de développement.
La première étape consiste à classifier le dispositif (classe I, IIa, IIb ou III) dès le départ. Cette classification détermine le niveau d'exigence réglementaire et les normes spécifiques à respecter. La classification est directement liée aux revendications sur le DM, c’est à ce moment-là qu’entrera en jeu la nation de stratégie réglementaire.
Ensuite, vous devez identifier toutes les normes applicables à votre dispositif : ISO 10993 pour la biocompatibilité si contact corporel, série IEC 60601 pour la sécurité électrique des équipements médicaux, normes spécifiques à certains types de dispositifs.
Les contraintes qui suivent doivent être intégrées dans les spécifications techniques :
C'est ici qu'intervient votre partenaire de développement en vous apportant son expertise normative dès la conception, en identifiant des contraintes non anticipées, en guidant les arbitrages techniques.
Au-delà de l'anticipation réglementaire, une approche méthodologique spécifique s'impose. Le concept de Design for Compliance prolonge naturellement l'approche DfMA (Design for Manufacturing & Assembly). Il s'agit de concevoir un produit qui soit simultanément fonctionnel, industrialisable et conforme aux exigences réglementaires.
La bill of materials illustre parfaitement cette triple contrainte. Ainsi, pour chaque matériau envisagé, vous devez vérifier non seulement ses propriétés mécaniques ou électriques, mais aussi la disponibilité de certificats de conformité tels que :
Dans la même logique, la conception modulaire facilite à la fois la traçabilité et les modifications futures. Un produit conçu en modules bien définis permet de tracer précisément l'origine de chaque sous-ensemble, de valider séparément chaque fonction, et de modifier un module sans remettre en cause l'ensemble de la validation.
Parallèlement, chaque décision de conception doit être documentée en temps réel, pas a posteriori. Cette rigueur quotidienne évite la reconstruction laborieuse de l'historique de projet. Enfin, les tests précoces s'inscrivent dans cette logique avec un prototypage itératif et des validations partielles à chaque étape.
Maintenant que votre produit est conçu, la certification peut prendre plusieurs mois selon la maturité organisationnelle et la préparation du système de management de la qualité. La roadmap passe par 6 étapes qu’il s’agira de bien préparer car c’est un marathon et non un sprint.
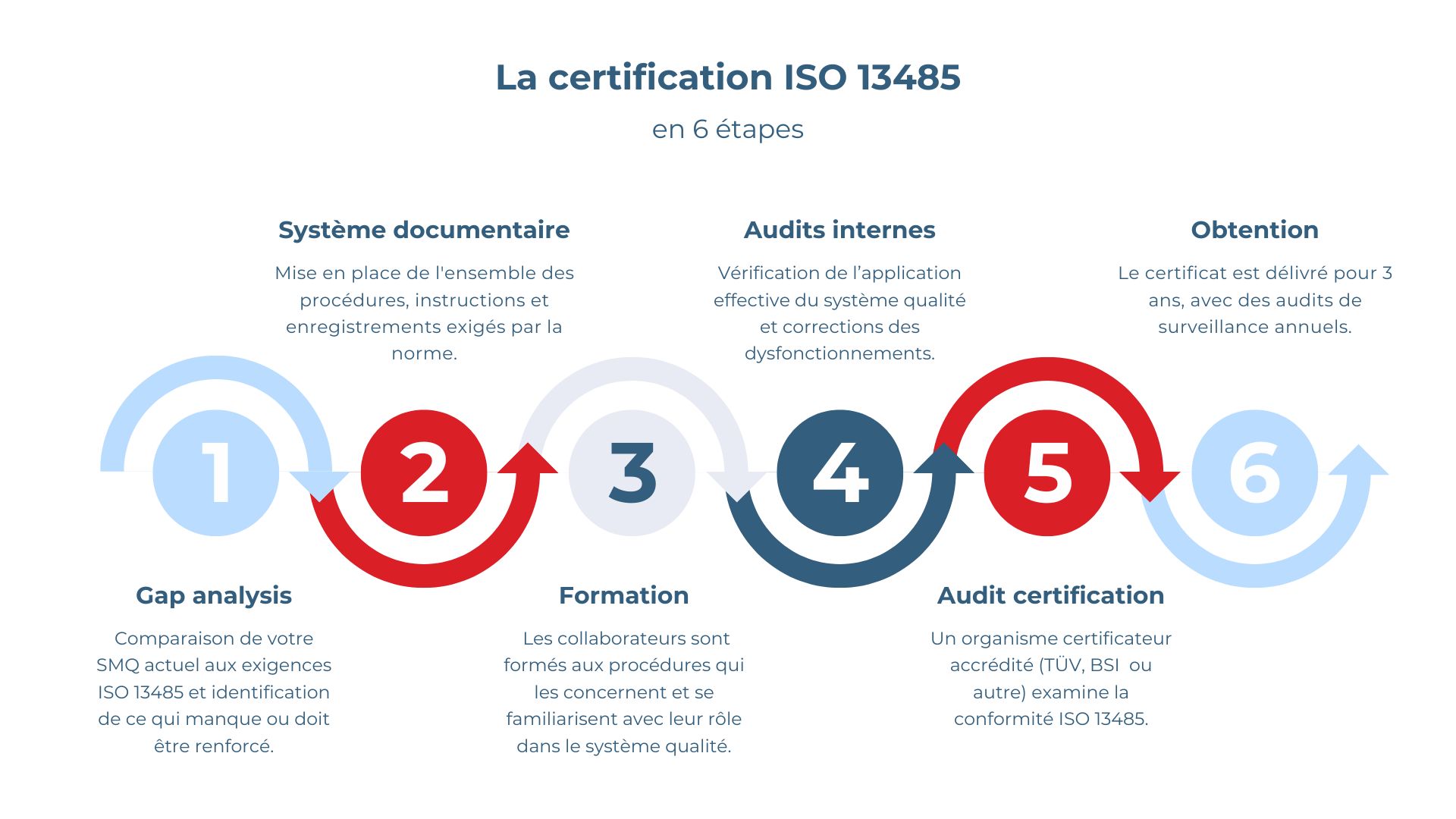
C’est contre-intuitif mais intégrer l'ISO 13485 dès le départ accélère le time-to-market au lieu de le ralentir. Plusieurs raisons à cet état de faits.
D'abord, vous évitez les refontes. Un prototype développé sans considération des exigences ISO 13485 révèle souvent des non-conformités majeures : matériaux non validés, impossibilité de tracer certains composants, absence de documentation prouvant les choix techniques. Corriger ces problèmes nécessite de refaire la conception et de recommencer les validations.
De plus, la documentation continue vous fait également gagner beaucoup de temps. Documenter au fur et à mesure du développement est plutôt rapide, alors que reconstituer a posteriori l'historique complet d'un projet peut s'avérer beaucoup plus long et mobiliser plusieurs ingénieurs.
Enfin, la validation progressive permet de résoudre les non-conformités étape par étape plutôt que de tout découvrir lors de l'audit final. Cette approche itérative réduit drastiquement le risque de mauvaise surprise tardive. In fine, l'acceptation marché est plus rapide : votre dossier technique est prêt dès la fin du développement.
La conformité ISO 13485 protège votre entreprise en réduisant les risques :
En vérité, la certification ISO 13485 constitue un avantage concurrentiel : elle différencie votre offre, rassure les clients professionnels (hôpitaux, cliniques, distributeurs et éventuels investisseurs) et ouvre l'accès aux marchés réglementés internationaux.
L’erreur sans doute la plus répandue, consiste à considérer ISO 13485 comme une case à cocher en fin de projet : "On développe d'abord notre produit comme on l'entend, on verra la certification après." En apparence pragmatique, cette logique séquentielle crée en réalité un fossé entre ce qui a été conçu et ce qui devrait l'être pour être conforme.
Les conséquences sont dès lors inévitables : découverte tardive de non-conformités majeures qui nécessitent de repenser des pans entiers de la conception. Ces refontes entraînent des retards importants et des surcoûts significatifs par rapport au budget initial.
Prenons un scénario type. Une entreprise développe pendant 18 mois un dispositif fonctionnel, puis découvre que les matériaux nécessitent une analyse de biocompatibilité en laboratoire. Résultat : délai complémentaire de laboratoire + potentielle reconception, 12 mois de retard.
À l'opposé, la bonne pratique consiste à faire de l'ISO 13485 un pilier du développement dès le kick-off du projet. La norme devient alors un cadre structurant qui guide les choix techniques, plutôt qu’une contrainte qu’on subit après coup.
La seconde erreur majeure, plus insidieuse, concerne l'application du système qualité. Un système de management de la qualité ISO 13485 n'existe que s'il est appliqué au quotidien par les équipes. La documentation la plus rigoureuse reste lettre morte si les collaborateurs ne la comprennent pas ou ne l'appliquent pas.
Concrètement, l'erreur classique consiste à rédiger des dizaines de procédures détaillées sans former les collaborateurs à leur utilisation. Ces documents finissent alors dans un classeur ou sur un serveur, mais n'irriguent pas les pratiques réelles.
Le risque qui en découle est double. D'une part, la non-application des procédures crée des écarts entre ce qui est écrit et ce qui est fait — écarts immédiatement détectés lors des audits et constituant des non-conformités majeures. D'autre part, sans application effective, le système qualité ne remplit pas sa fonction première : garantir la qualité et la sécurité du dispositif.
Pour éviter cet écueil, l'acculturation qualité doit toucher chaque membre de l'équipe. Il ne s'agit pas seulement de connaître les procédures, mais de comprendre le "pourquoi" : pourquoi cette exigence de traçabilité, pourquoi cette validation, pourquoi cette documentation. Cette compréhension transforme la contrainte perçue en pratique naturelle.
Par ailleurs, il est essentiel de mettre en place un Système de Management de la Qualité (SMQ) simple et facile d'utilisation, conçu et déployé par un expert. La norme ISO 13485 ne dicte pas la manière exacte de structurer un SMQ ; il convient donc d'interpréter ses exigences pour créer un système qui soit conforme à la norme tout en étant adapté aux besoins spécifiques du fabricant de dispositifs médicaux.
Dans cette dynamique, si la direction n'incarne pas elle-même la culture qualité, si elle tolère les raccourcis ou les dérogations, tout le système s'effondre. La qualité est une affaire de cohérence entre discours et pratique.
En résumé, dans un secteur où les enjeux réglementaires se renforcent continuellement, la maîtrise de l'ISO 13485 n'est plus optionnelle. Elle constitue un différenciateur stratégique qui sépare les acteurs capables d'industrialiser leurs innovations de ceux qui restent au stade du prototype.
L'ISO 9001 est une norme généraliste de management de la qualité qui s'applique à tous les secteurs. L'ISO 13485 est spécifiquement conçue pour les dispositifs médicaux et va beaucoup plus loin sur trois aspects : la traçabilité complète de chaque composant et lot, la gestion formalisée des risques selon ISO 14971, et des exigences documentaires beaucoup plus strictes.
La durée dépend de la maturité de votre organisation au départ. Le processus complet pour mettre en place le système documentaire, former les équipes, réaliser les audits internes et obtenir la certification peut prendre plusieurs mois. Une préparation insuffisante se traduit par des non-conformités lors de l'audit, ce qui retarde l'obtention du certificat.
Tous les dispositifs médicaux sont concernés, quelle que soit leur classe de risque (I, IIa, IIb, III). Le niveau de rigueur augmente avec la classe de risque. Même les fournisseurs et sous-traitants qui ne fabriquent qu'un composant du dispositif final doivent souvent démontrer leur conformité ISO 13485.
Découvrez l’intégralité de notre blog pour rester informé des tendances et expertises techniques qui font la force de Scalea.


